Two Examples of Parallel Computing in Materials Modeling
Dept. of Nuclear Engineering
Both authors work in the multiscale
materials modeling group under Prof. Sidney Yip in the Dept. of Nuclear Engineering
at MIT. The main research interest of our group is trying to
understand and predict material properties from basic physical
principles by doing numerical simulations and modeling, on length
scales spanning angstroms to microns. Parallel computing in recent
years has become a major thrust to this field and is quickly changing
the landscape of our research. As starters, we would like to be
convinced of its power by doing two relevant examples that are
important parts of our ongoing research. The machine we are going to
use is MIT's Xolas network of nine eight processor (SUN Ultra
Enterprise 5000) SMP's at the
Laboratory for Computer Science. Our motto is: ``For large scale
simulations speed is highly desirable if it's attainable''. A general
purpose parallel environment such as MPI, PVM, or Cilk often compromises speed to
generality and simplicity, so using them could not attain peak
performance. We choose to use the most primitive method, that is, by
making UNIX system calls (shmop,
fork, semop), as
our only parallelization technique, and shall demonstrate its high
performance on a shared memory SMP. The two examples are:
-
Parallelize an existent order-N code which calculates the local
density of states (LDOS) of large (rank ~ 10^4) sparse matrices
such as the dynamical matrix or tight-binding Hamiltonian occurring
in condensed-matter physics. Uses shared memory. For details
click here.
-
An order-N, parallel molecular dynamics code for pairwise
two-body interatomic potential with finite range, which can handle
10^5 atoms. Uses shared memory within one SMP. This code shall be
used to study the coupling method between continuum and MD
simulations. For details
click here.
The reason we put the two examples together is for comparison. The
first case is relatively simple: in its serial version (f77) the
program spends 90 percent of the time in a few loops which are
basically (complex) sparse matrix -- vector multiplications. We would
like to retain the f77 efficiency in handling complex number
operations (an intrinsic type), but it is very difficult to do any
kind of shared memory, multi-process programming using f77 as (almost
all) the memory addressing is static. We get around this problem by
doing hybrid coding with C: shared-memory
allocation, management and process synchronization is done in C,
but a few computationally intensive kernels were written as f77
subroutines -- the data in shared memory is passed to them as
arguments, which solved the memory addressing problem. In a sense our
method is a ``quick and dirty'' solution to parallelize a straight
forward f77 code while retaining most of the efficiency: this was
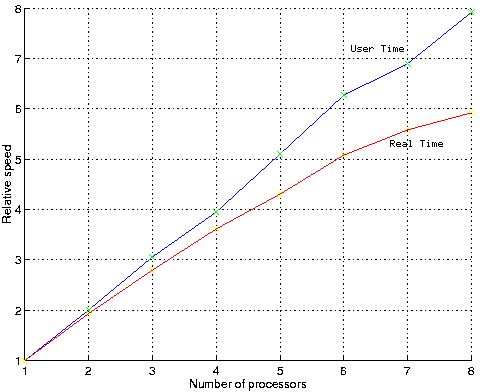
shown by the nearly ideal speed scaling plot
. The primitive master-slave communication mechanism through an
array of ``schedule flags'' in shared memory has almost no overhead in
this case, and because only the master process passes through the main
loop it is possible to do simple timing and speed benchmark. The drawbacks are 1) all the
parallelism must explicitly come from the human mind, which is error
prone. 2) The UNIX system call fork()
is the simplest way we can find to launch multi processors
to run on Xolas instead of (what we doubt to be)
multi processes on a single processor. But it has a large context and
performance isn't always stable. 3) For expediencies in programming
we had assumed the shared memory to be always coming in one block. In
actuality that is not always possible.
In the second example, a powerful molecular dynamics
code was developed for shared memory SMP. O(N) complexity can be
achieved by dividing the simulation cell into many bins: each bin
keeps a particle list and each particle keeps a neighbor list. One
processor can control many bins, the layout is generated on a
geometrical basis. A ``flash'' condition is devised for updating the
neighbor list of atoms. Maintaining the neighbor lists makes force
evaluation O(N), and maintaining the bin lists makes updating the
neighbor list O(N). Data dependency between processors includes force
evaluation, neighbor list maintainance and particle transfer near the
interface. In general, interface crossing happens frequently. Using MPI or
PVM would be too expensive, even inside a SMP. We used shared
memory directly: in our program all data are initialized in shared
memory and all processors access shared memory directly, not a single
cycle will be spent on data transfer! We use
semaphore to synchronize between processes and to maintain data
coherence, the benefit is that no resources will be wasted in empty
loops.
The serial version of the program was written in Fortran
90. In order to best utilize shared memory and semaphore, the code
was converted to C by hand, and by using many optimization
techniques which Fortran discourages, the converted code runs even
faster than the original F90 code on single processor.
Again, linear scaling of speed with the number of processors is
achieved as expected.
This work was done as part of the final project for 18.337/6.338 Parallel
Scientific Computing Spring 1997, taught by Prof. Alan Edelman. We would
like to thank the
Laboratory for Computer Science at MIT for providing
valuable computer time on the Xolas cluster.
Constructed and maintained by Li Ju (lij@matsceng.ohio-state.edu) and Liao Dongyi, (liaody@mit.edu).
Last modified on May. 14,1997. This page has been accessed
at least  times
since May. 14,1997.
times
since May. 14,1997.